Fast, accurate antibody structure prediction from deep learning on massive set of natural antibodies
Abstract
Antibodies have the capacity to bind a diverse set of antigens, and they have become critical therapeutics and diagnostic molecules. The binding of antibodies is facilitated by a set of six hypervariable loops that are diversified through genetic recombination and mutation. Even with recent advances, accurate structural prediction of these loops remains a challenge. Here, we present {IgFold}, a fast deep learning method for antibody structure prediction. {IgFold} consists of a pre-trained language model trained on 558 million natural antibody sequences followed by graph networks that directly predict backbone atom coordinates. {IgFold} predicts structures of similar or better quality than alternative methods (including {AlphaFold}) in significantly less time (under 25 s). Accurate structure prediction on this timescale makes possible avenues of investigation that were previously infeasible. As a demonstration of {IgFold}’s capabilities, we predicted structures for 1.4 million paired antibody sequences, providing structural insights to 500-fold more antibodies than have experimentally determined structures.
Notes:
- Five of the existing CDR loops can be predicted effectively by sequence similarity, however, CDR-H3 is more diverse in sequence and length than the other CDRs. Additionally, CDR-H3 is at the interface between the heavy and light chains, making its conformation dependent on the inter-chain orientation.
- Leverages embeddings from AntiBERTy.
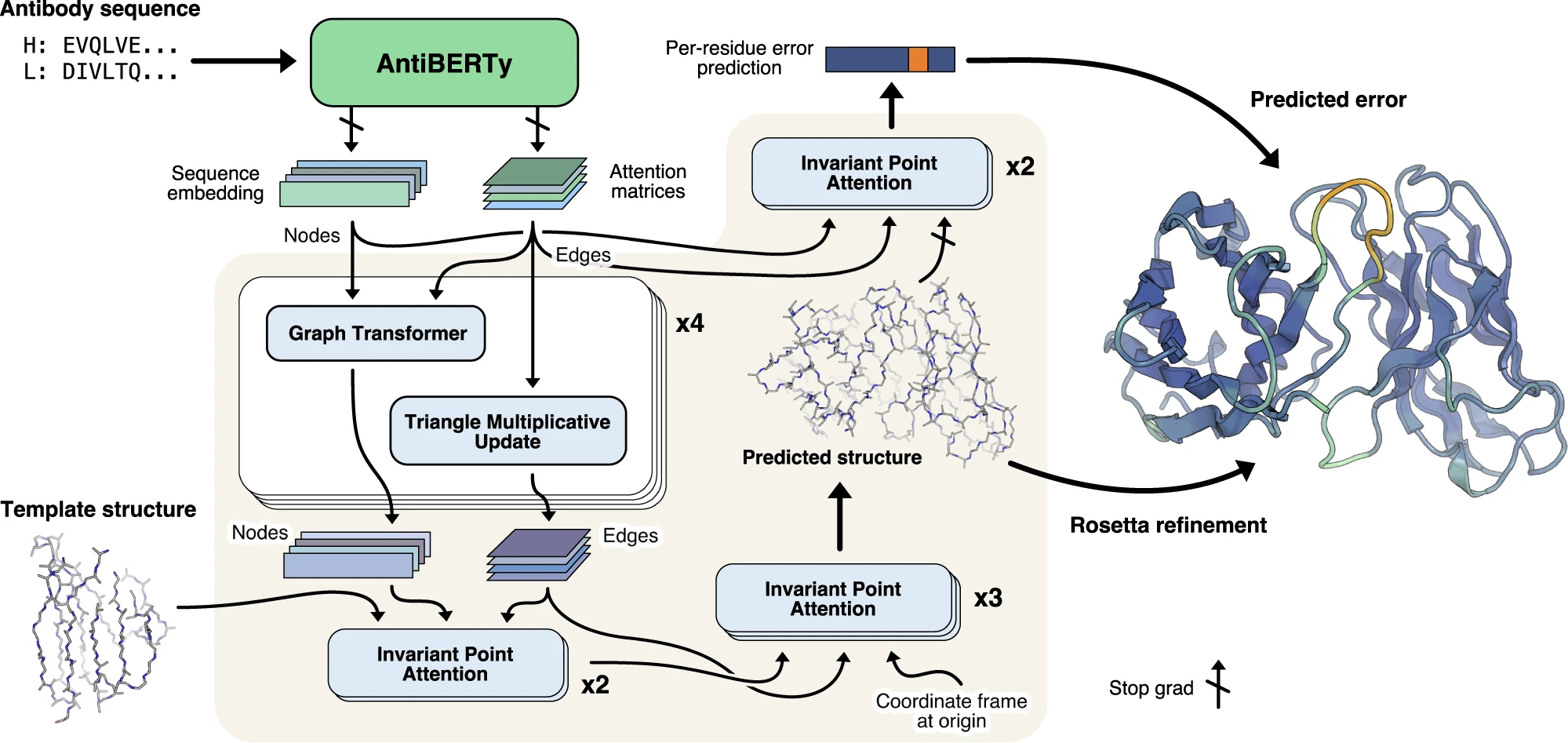
- Invariant point attention prevents the embedding from changing based on structure orientation
- Template coordinates are fixed
- MSE of predicted to experimental coordinates was used as the loss value
Quote
Rather than search for structural templates for training, we generate template-like structures by corruption of the true label structures. Specifically, for 50% of training examples, we randomly select one to six consecutive segments of twenty residues and move the atomic coordinates to the origin. The remaining residues are provided to the model as a template. The deleted segments of residues are hidden from the IPA attention, so that the model only incorporates structural information from residues with meaningful coordinates.
Comment: Is this a fair way to train the model / Does it generalise well? This is not how the model is used in real life, templates will not have 100% seq id with the requested structure.
Quote
Predicting structures for such heavily engineered sequences is challenging, particularly for models trained primarily on natural antibody structural data (such as IgFold). To investigate whether IgFold’s error estimations can identify likely mistakes in such sequences, we predicted the structure of an anti-HLA (human leukocyte antigen) antibody with a sequence randomized CDR H1 loop. As expected, there is significant error in the predicted CDR H1 loop structure. However, the erroneous structure is accompanied by a high error estimate, revealing that the predicted conformation is likely to be incorrect. This suggests that the RMSD predictions from IgFold are sensitive to unnatural antibody sequences and should be informative for a broad range of antibody structure predictions.
Comment: I don’t think we can determine IgFold is sensitive to unnatural antibody sequences from one randomised example. Is it accurate when predicting engineered antibodies that aren’t a randomised sequence?
- Predicted RMSD values tend to be underestimations, most likely due to the class imbalance (more low RMSD FR residues than high RMSD CDR residues).
Question
Instead of training from AntiBERTy could you fine tune AF-M on antibody structures?
Abstract
- Does Repetoire Builder represent Schrodinger and MOE’s homology modelling performance?
- Comparison between DL & Repertoire builder might not be fair - Rosetta refinement is run for IgFold and other DL models but not RB (Could Rosetta just be that good?), however, RB reported only minor RMSD improvements with Rosetta so this point could be irrelevant. RB instead uses SCWRL4 which only alters side chains and not bond length / angles.
- Could run benchmark dataset through schrodinger and MOE.
- Engineered sequences deviate from natural antibody sequences that IgFold is trained on, does homology modelling still perform okay on engineered seqs? The provided example for IgFold showed it performing poorly when predicting an Anti-HLA antibody structure with randomised CDRH1.
Metadata:
Type:&Tags:#🛸Author:Jeffrey RuffoloKeywords:#antibody_modelling,#deep_learningDOI:DOIZotero URL:ZoteroReviewed Date:2024-12-27
Citation
@article{ruffolo_fast_2023,
title = {Fast, accurate antibody structure prediction from deep learning on massive set of natural antibodies},
volume = {14},
issn = {2041-1723},
url = {https://www.nature.com/articles/s41467-023-38063-x},
doi = {10.1038/s41467-023-38063-x},
pages = {2389},
number = {1},
journaltitle = {Nature Communications},
shortjournal = {Nat Commun},
author = {Ruffolo, Jeffrey A. and Chu, Lee-Shin and Mahajan, Sai Pooja and Gray, Jeffrey J.},
urldate = {2024-12-27},
date = {2023-04-25},
langid = {english},
file = {Full Text:/Users/mikerophone/Zotero/storage/PY3VRV8W/Ruffolo et al. - 2023 - Fast, accurate antibody structure prediction from deep learning on massive set of natural antibodies.pdf:application/pdf},
}